cardiopatias congenitas


El corazón es el primer órgano que se forma y funciona en el embrión como un tubo primitivo, de tal forma que todos los eventos subsecuentes en la vida de un organismo dependen de su funcionalidad. Las mutaciones heredadas en los genes que intervienen en el desarrollo cardioembrionario, pueden provocar una enfermedad cardíaca congénita, que es la forma más común de defectos humanos del nacimiento (1% de todos los nacimientos), o anormalidades en el corazón adulto que son la causa más prevalente de morbi–mortalidad en el mundo industrializado.12 En los últimos diez años, se ha observado una marcada transición en los estudios fisiológicos del corazón, de tal suerte que se ha avanzado mucho en el conocimiento de la disfunción cardíaca a nivel genético y molecular. Estos avances científicos y tecnológicos han dado la oportunidad de crear nuevas aproximaciones terapéuticas para la prevención y el tratamiento de enfermedades cardíacas y, además, han generado nuevas incógnitas a resolver en este tema, provocando cambios y nuevas oportunidades en el desarrollo de terapias para las enfermedades cardíacas congénitas y adquiridas.
El entendimiento de los mecanismos del desarrollo cardíaco provee de nuevos conocimientos en los fenotipos cardíacos anormales. El desarrollo de los organismos transgénicos ha permitido reproducir defectos cardíacos gobernados por un solo gen (monogénicos) el cual se depleta o inhabilita en forma homocigota, aunque la mayoría de las mutaciones que causan enfermedad cardíaca en el humano se presentan en forma heterocigota. Por otro lado, los defectos cardíacos congénitos en el humano presentan una gran variabilidad en cuanto a su pene–trancia y expresividad, lo cual indica que, además del efecto que causa el o los genes mutados, las influencias medioambientales también participan en el fenotipo de la enfermedad.3
Los avances recientes en genética molecular han revelado que existen factores genéticos y moleculares ligados a enfermedades cardíacas congénitas y arritmias cardíacas, y han permitido su identificación en el mapa genómico (Fig. 1), con lo cual se ha creado una invaluable oportunidad para innovar diagnósticos genéticos y, en un futuro, alcanzar la terapia génica.

I. Cardiopatía congénita:
Existe una gran variedad de defectos congénitos asociados a mutaciones simples y dichas mutaciones se presentan en un amplio espectro de genes involucrados en la estructura y funcionamiento cardíaco. El nivel para la especificidad cardíaca en estas mutaciones es altamente variable. Algunos genes mutados se asocian a síndromes con una presentación neuromuscular sistémica que también involucra al corazón (por ejemplo: Ataxia de Friedreich y Distrofia muscular de Duchenne). Un amplio rango de defectos cardíacos resulta de estas mutaciones genéticas, incluyendo anormalidades en la función electrofisiológica (defectos de la conducción y arritmias), proteínas de la matriz extracelular, enzimas y transportadores de membrana involucradas en la biosíntesis de ácidos grasos y función mitocondrial, metabolismo de la fosforilación oxidativa, estructura sarcomérica, proteínas contráctiles y factores de transcripción nuclear que regulan la expresión genética miocárdica y su desarrollo programado (Tabla I).
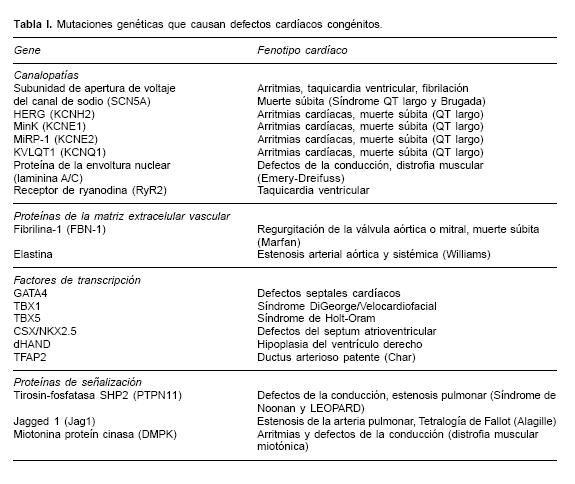
Las malformaciones cardíacas pleiotropicas pueden ser el resultado de mutaciones discretas en factores de transcripción específicos y proteínas clave en el desarrollo embrionario y la morfogénesis cardíaca.4–6 Factores como GATA4, NKX2.5, dHAND, TFAP2 y TBX5, son genes que se expresan tempranamente durante el desarrollo embrionario cardíaco y su expresión es crucial para la activación de otros genes cardíacos. Mutaciones específicas en cada uno de estos genes representan severas anormalidades cardíacas como: defectos septales (GATA4), defectos de la conducción (NKX2.5), hipoplasia ventricular derecha (dHAND), ductus arteriosus patente en el síndrome de Char (TFAP2B) y síndrome de Holt–Oram (TBX5), entre otros, cuya acción es crítica en la disrupción del desarrollo cardíaco temprano y la morfogénesis cardíaca dentro de la génesis de los defectos congénitos cardíacos.7–11
Así mismo, existen defectos genéticos en proteínas involucradas en múltiples rutas de señalización que modulan la proliferación, migración y diferenciación celular en la embriogénesis cardíaca. Se han detectado mutaciones en el gen Jag1en estudios de asociación con el síndrome de Alagille, un desorden autosómico dominante que se presenta con defectos congénitos cardíacos que incluyen estenosis de la arteria pulmonar y tetralogía de Fallot.12
El gene PTPN11 codifica para la tirosin fosfatasa (SHP–2), de tal suerte que mutaciones en este gen pueden causar síndrome de Noonan, el cual se caracteriza por defectos de la conducción, estenosis pulmonar, cardiomiopatía hipertrófica13 y recientemente se le ha involucrado en la patogénesis del síndrome de LEOPARD (lentígines múltiples, electrocardiograma anormal, hipertelorismo, estenosis de la arteria pulmonar, anormalidades genitales, retraso en el crecimiento y sordera) el cual puede ser un desorden alélico.14
Además de las mutaciones puntuales, en regiones codificantes (exones) de genes específicos, una gran cantidad de desórdenes neuromusculares heredados se han referido como síndromes de triple repetición, en los cuales está incluida la ataxia de Friedreich y la distrofia muscular miotónica, que son causadas por secuencias trinucleotídicas repetidas en genes específicos, por ejemplo en el gen de la frataxi–na (FRDA) y el de la miotonin proteína cinasa (DMPK), respectivamente.15,16 Los individuos afectados presentan cardiomiopatía, arritmias cardíacas y defectos en la conducción. En ambos defectos, la severidad de la enfermedad correlaciona con el número de tripletes repetidos, por ejemplo: individuos con ataxia de Friedreich presentan > 200 repetidos GAA, mientras que en la distrofia muscular miotónica, los individuos afectados presentan más de 50 copias del triplete CTG.
Otro tipo de defectos genéticos involucrados en malformaciones del desarrollo estructural cardíaco, son las grandes deleciones o microdeleciones cromosomales que provocan anormalidades conotroncales, defectos del canal atrioventricular y defectos del septum atrial y ventricular.17,18 Defectos del tracto de salida cardíaco son manifestaciones de un desorden genético complejo denominado Síndrome DiGeorge/velocardiofacial o CATCH22 (defectos cardíacos, facies anormales, hipoplasia tímica, paladar hendido, hipocalcemia). La mayoría de los pacientes son hemicigotos para una microdeleción que abarca una región de 1.5 a 3 megabases en el cromosoma 22 (22q11.2), la cual es esencial para el desarrollo normal del arco faríngeo. La deleción del 22q11, es un evento relativamente común que se da en 1 de cada 4,000 nacidos vivos. Se piensa que el gene TBX1, ubicado en la parte central de la región deletada, es un factor crítico en el desarrollo de este defecto congénito.19 Dicho gen codifica para un factor de transcripción involucrado en la regulación del desarrollo cardíaco y es miembro de una familia de genes altamente conservados que comparten un dominio de unión a DNA (el T–box). La reducción de la expresión de TBX1 (la cual ocurre en estado hemicigoto), representa una haploinsuficiencia que repercute ampliamente en la expresión de varios genes de expresión temprana en la morfogénesis cardíaca. Se han reportado otras microdeleciones cromosómicas asociadas a defectos cardíacos congenitos (8p) y es posible que se localicen otras más.20 Las técnicas citogenéticas moleculares actuales presentan una alta resolución, como el caso de la hibridación fluorescente in situ (FISH), que se utiliza rutinariamente para confirmar el diagnóstico clínico de daño cromosómico como en el caso de microdeleciones y pequeñas translocaciones.
Es importante hacer notar que las grandes deleciones genéticas se asocian comúnmente con un amplio espectro de características clínicas adicionales al daño cardíaco. Las malformaciones extracardíacas se asocian frecuentemente con defectos cardíacos congénitos y su incidencia se ha calculado en un 30% de los casos. Dichas anomalías cromosómicas son más prevalentes en individuos con defectos cardíacos que en la población general. Estas malformaciones cardíacas son el resultado de aberraciones cromosómicas como lo es el caso de las trisomías 13, 18 y 21 (Síndrome de Down), así como de la monosomía X0 (Síndrome de Turner), cuyas bases moleculares precisas aún no han sido del todo elucidadas.
II. Arritmias y muerte súbita:
Las arritmias cardíacas son complicaciones frecuentes de enfermedad cardíaca pediátrica y pueden ser la principal causa de muerte súbita. Se han identificado mutaciones en genes que codifican para canales iónicos cardíacos, como factores de riesgo en la patogénesis de arritmias letales y no letales.
El gen SCN5A codifica para un canal de sodio responsable del inicio del potencial de acción. Mutaciones en este gen se asocian a la prolongación del intervalo QT o Síndrome de QT largo (LQTS) el cual predispone al síncope y muerte súbita.21,22 Su fenotipo es una repolarización ventricular anormal que puede resultar en fibrila–ción ventricular idiopática, taquicardia ventricular, defectos de la conducción cardíaca y síndrome de Bragada.23,24 Existen 4 genes más involucrados en la formación de canales de potasio que también se han asociado al LQTS, ellos son el HERG, KCNE1, KCNE2 y KVLQT1 y en ellos se han descrito más de 30 diferentes mutaciones estudiadas en 40 familias con gran heterogeneidad genética.25
Se han identificado mutaciones de sentido erróneo (missense) en el gen del receptor rianodin (RyR2), que es un canal liberador de calcio involucrado en la contracción–relajación sarcomérica, y que causa estrés por sobrecarga de calcio en miocitos, lo cual provoca taquicardia ventricular.26 Algunas mutaciones en el gen de lamini–na A/C, que codifican para proteínas de la envoltura nuclear, se presentan en individuos afectados por la forma autosómica dominante de la distrofia muscular de Emery Dreifuss, en la que se observa lipodistrofia parcial familiar, car–diomiopatía dilatada (DCM), defectos de la conducción atrioventricular y fibrilación atrial.27 Por otro lado, la acumulación de metabolitos intermediarios de los ácidos grasos, como la cadena larga de acilcarnitina, pueden provocar arritmias cardíacas y defectos en la conducción en neonatos. Se han reportado errores del nacimiento en la oxidación de ácidos grasos (por ejemplo, deficiencias en la carnitin:palmitoil–transferasa II, proteína trifuncional mitocondrial o en la carnitil–acil–carnitin translocasa), en la muerte súbita infantil inexplicable y en infantes con defectos de la conducción o taquicardia ventricular.28
II. Vasculopatías:
Dentro de las vasculopatías, consideradas como defectos autosómicos dominantes, se encuentra el síndrome de Marfán, la estenosis aórtica supravalvular y el síndrome de Williams, en los cuales se evidencia el papel crítico de las microfibrillas, y defectos de la matriz extracelular en la patofisiología de estos defectos génicos.29,30 El síndrome de Marfán se caracteriza por anormalidad de los sistemas músculo–esquelético y cardiovascular, además de los ojos, que llevan a una muerte prematura principalmente por una dilatación progresiva del tronco aórtico con una disección aórtica fatal o por insuficiencia valvular aórtica y se asocia con una gran mortalidad neonatal con participación polivalvular y falla cardíaca congestiva severa. La mayoría de los casos con síndrome de Marfán y enfermedad cardiovascular, tienen mutaciones en el gen de la fibrilina y la mayoría de sus familiares presentan diferentes mutaciones ahí. La fibrilina es un constituyente de un complejo multiproteico (que incluye a la elastina) presente en el componente microfibrilar de la pared de los grandes vasos. Mutaciones en los genes que codifican para los componentes de la matriz extracelular (por ejemplo: elastina), son responsables de la estenosis supravalvular aórtica cuyas características obstructivas afectan a la aorta ascendente. El síndrome de Williams se caracteriza por presentar estenosis de las arterias pulmonares o sistémicas.
IV. Cardiomiopatía:
Las mutaciones que causan cardiomiopatías en el humano se han identificado en un amplio espectro de genes nucleares que codifican para proteínas contráctiles y estructurales, enzimas involucradas en el almacenamiento del glucógeno (enfermedades de Cori y Pompe) y degradación de mucopolisacáridos (enfermedad de Fabry), metabolismo de lípidos (β–oxidación de ácidos grasos y deficiencia de la carnitina) y en los genes esenciales para la producción de energía cardíaca (Tabla II), ubicados en el DNA nuclear y mitocondrial (mtDNA). Se ha observado que tanto la cardiomiopatía dilatada (DCM) como la cardiomiopatía hipertrófica (HCM) se presentan en jóvenes y pueden tener componentes genético–familiares. La HCM se ha estudiado más ampliamente debido a que representa la causa más frecuente de muerte súbita en niños y adolescentes. La mayoría de casos familiares de HCM presentan una forma de transmisión autosómica dominante, a excepción, claro, de aquellos casos con mutaciones en el mtDNA, las cuales son heredadas por vía materna (Fig. 2). Se han observado más de 10 genes involucrados en la HCM que codifican para proteínas de la sarcómera como la cadena pesada de α y β–miosina (α y β–MHC), proteína C de unión a miosina, troponina I y troponina T, α–tropomiosina, cadenas ligeras de miosina esencial y regulatoria, titina y α–actina.31–41 También se han detectado mutaciones en pacientes con HCM en genes involucrados en el metabolismo del grupo hemo y Fe++ (Frataxina y COX15)15,37 y en la bioenergética mitocondrial (genes mitocondriales para tRNAs y ATPasa6).38 Un subgrupo de casos conHCM mostró mutaciones en la subunidad reguladora de la proteína cinasa de AMP (AMPK), la cual es un sensor clave y mediador del metabolismo energético.39 En resumen, todos estos resultados sugieren que la interrupción del metabolismo energético mitocondrial cardíaco es la causa de HCM en aquellos pacientes con problemas de contracción sarcomérica y nos pueden ayudar a entender numerosas observaciones clínicas como su heterogeneidad, variabilidad de presentación clínica y su asimetría hipertrófica.


Actualmente, se estima que aproximadamente el 30% de los casos con DCM son heredados, y el 70% son casos esporádicos. Se han identificado genes relacionados con la DCM familiar ligada al cromosomaX (Distrofina, G4.5)40,41 y con la forma autosómica dominante de DCM (acuna, desmina, laminina A/C, δ–sarcoglicano).42–44 En el caso de la DCM ligada al cromosoma X, el gen de la distrofina (enorme proteína del citoesqueleto asociada a la sarcómera) se ve afectado sólo en los miocitos, de tal suerte que las mutaciones que lo afectan se ubican en su promotor tejido específico.45 Mutaciones directas sobre el gen de la distrofina provocan distrofia muscular de Duchenne o de tipo Becker (DMD, DMB), en las cuales se afectan tanto el músculo cardíaco como el esquelético en general.40 La forma más grave de la DMD es provocada por mutaciones o grandes deleciones que afectan el marco de lectura traduccional o aquellas mutaciones puntuales que crean codones de paro, que afectan la producción completa de la proteína. Los varones afectados en la DMD son asintomá–ticos al principio de su adolescencia, pero desarrollan síncope y falla cardíaca congestiva rápidamente progresiva en el final de su adolescencia; las mujeres generalmente presentan la sintomatología más tardíamente. En este tipo de distrofia se debilitan los músculos esqueléticos a edad temprana (3 a 6 años) y posteriormente más del 30% de los casos presentan disfunción cardíaca alrededor de los 14 años; virtualmente, todos los pacientes con DMD desarrollan DCM a los 18 años.
El síndrome de Barth, una miopatía cardioesquelética ligada al X, con neutropenia y cardio–miopatía dilatada que se presenta en la infancia, es producido por mutaciones del gen G4.5 que codifica para la tafacina, una proteína de la familia de aciltransferasas involucrada en la síntesis de fosfolípidos.41–46 Los pacientes con este síndrome presentan niveles elevados de ácidos grasos saturados y muy bajos de cardiolipina, lo cual afecta la fluidez de la membrana de los cardiomiocitos y su función. La displasia arritmogénica del ventrículo derecho (ARVD) se presenta en forma autosómica dominante caracterizada por degeneración progresiva del miocardio, arritmias y gran riesgo de muerte súbita. El análisis de ligamiento ha mostrado que este desorden involucra varios loci de los cromosomas 2, 10, 14 y 17, pero el sitio exacto del defecto genético no se ha determinado.47
Se han descrito varias cardiopatías mitocondriales asociadas a desórdenes neurológicos como el MELAS (miopatía mitocondrial, encefalopatía, acidosis láctica y episodios convulsivos), el MERRF (epilepsia mioclónicay fibras rojas rasgadas) y el síndrome de Leigh38,48 en genes del mtDNA y también en genes nucleares involucrados en el ensamble de la cadena respiratoria mitocondrial (Fig. 2). Estos desórdenes pueden presentarse de forma temprana o tardía y causar cardiomiopatía infantil fatal.49–51 Los estudios moleculares de pacientes con HCM o DCM han aportado gran información para la identificación de nuevas mutaciones en el mtDNA que prevalecen en el tejido cardíaco.52,53 Estas mutaciones también se han encontrado recientemente en genes que codifican para los complejos respiratorios mitocondriales, I, III, y IV, además del gen ATP6 de la F1F2–ATP sintasa (Tabla II). Otros genes nucleares involucrados en la biogénesis mitocondrial o en el ensamblaje de los complejos respiratorios, también son blancos de mutaciones que provocan cardiomiopatías; tal es el caso de SCO2 que controla el ensamblaje de la citocromo oxidasa (Complejo IV), o los genes NDUFS2 y NDUSF4 que son parte estructural de la NADH deshidrogenasa o complejo I mitocondrial.54 Las cardiomiopatías mitocondriales pueden ser causadas por eventos esporádicos como lo son los agentes que causan daño al mtDNA como el alcohol y la adriamicina.55,56 Las mutaciones somáticas así generadas pueden incrementarse durante la isquemia miocárdica y generar daño por estrés oxidativo.55-59 El síndrome de Kearns–Sayre, un desorden neuromuscular con defectos de la conducción atrioventricular y cardiomiopatía, se asocia con deleciones abundantes del mtDNA que se cree que son provocadas de forma espontánea porque no se detectan en la madre o hermanos del afectado.60
En contraste, la DCM se ha asociado a múltiples deleciones en el mtDNA y se pueden presentar de manera dominante o recesiva.61 Las familias que presentan una herencia dominante presentan mutaciones en proteínas necesarias para la replicación del mtDNA, como el gene que codifica para la mtDNApol γ y el gene Twinkle que produce una helicasa, o el metabolismo nucleotídico mitocondrial, como el translocador nucleotídico de adenina.62 Las mutaciones autosómicas recesivas se presentan en factores que juegan un papel importante en el metabolismo nucleotídico mitocondrial, como la timidincinasa 2, timidin–fosforilasa y deoxy–guanosin–cinasa.62 Además, las mutaciones mitocondriales pueden ser inducidas también por zidovudine (AZT), el cual inhibe la acción de las DNApol virales y mitocondriales, con lo cual afecta a la replicación,63 aunque aún no se conoce que este compuesto afecte en el desarrollo de cardiomiopatía en niños tratados con AZT.64
Técnicas de diagnóstico molecular, limitaciones y avances:
Muchos de los genes nucleares implicados en las cardiopatías fueron mapeados por análisis de ligamento en las familias afectadas y esto permitió su posterior identificación como genes candidatos por clonación posicional y secuencia–ción. Una gran variedad de técnicas moleculares, incluyendo la reacción en cadena de la polimerasa (PCR), restricción de fragmentos polimórficos (RFLP's) y polimorfismos conformacionales de cadena sencilla (SSCP), se han utilizado para rastrear alelos defectuosos del caso índice y sus familiares para establecer los patrones de herencia. En la mayoría de los casos, la detección de nuevas mutaciones se realizó sobre las secuencias codificantes (exones) de los genes candidatos y los casos relativamente bien caracterizados de HCM familiar han mostrado que las mutaciones que provocan la enfermedad son casos raros o mutaciones "de novo" en estas familias. Así pues, una correlación genotipo–fenotipo como en mutaciones específicas de β–MHC en la HCM se pueden asociar con una alta incidencia de muerte súbita, mientras que otras mutaciones se asocian con un mejor pronóstico. Recientes avances en la velocidad y sensibilidad de la detección de mutaciones aplicando técnicas como la cromatografía líquida altamente desnaturalizante (DHPLC) o la electroforesis capilar de alta resolución, pueden potenciar el uso de los análisis genéticos–moleculares para establecer un diagnóstico preclínico o clínico y proveer tratamientos en blancos específicos de desordenes cardíacos pediátricos. Así mismo, en un futuro cercano, la disponibilidad de la tecnología a base de chips genéticos permitirá un rastreo rápido y automatizado de mutaciones en el DNA nuclear y mitocondrial.
Desde que las técnicas modernas de imagenología han ayudado a determinar los fenotipos cardíacos en niños afectados, la heterogeneidad genética y la variabilidad intrafamiliar han precisado la elucidación molecular de muchos defectos cardíacos, así como la correlación genotipo–fenotipo que regularmente es difícil de observar. Estas dificultades pueden atribuirse a factores poligénicos o multifactoriales que contribuyen a la expresión de defectos cardíacos específicos, así como también a una gran cantidad de influencias epigenéticas o adquiridas. El progreso es gradual y comienza a definir estos factores epigenéticos y poligénicos, algunos de los cuales son afines al análisis molecular.
Se han realizado estudios de asociación de polimorfismos de bases simples en donde se han detectado genes candidatos para varios padecimientos cardíacos como infarto del miocardio, enfermedad de arterias coronarias y cardiomiopatía hipertrófica (Tabla III). Estos estudios han generado bases de datos que se encuentran disponibles para evaluar los efectos de los polimorfismos en la predisposición a defectos cardíacos específicos y quizá tengan impacto en las opciones de diagnóstico y tratamiento. Los recientes avances en las metodologías han hecho posible la realización de análisis detallados de la expresión genética con el uso de muestras pequeñas de tejido, lo cual es muy importante en el estudio de neonatos y niños. Una de estas metodologías se basa en los microarreglos de DNA (Microarrays). Estos microarreglos son constructos artificiales de DNA arreglados en forma de rejillas (hileras y columnas) en el cual cada elemento ubicado en cada una de las celdas de la rejilla, actúa como una sonda para hibridar a un RNA específico. La expresión génica por análisis de microarreglos ofrece una potente herramienta para establecer las características patofisiológicas de una enfermedad por la evaluación del incremento o decremento de expresión genética, pudiendo aplicarse al diagnóstico y a la evaluación de tratamientos aplicados a los pacientes.

La asociación de genes defectuosos con desórdenes cardíacos no cubiertos por análisis genómicos, necesitan ser seguidos por análisis pro–teómicos para establecer la función y papel patofisiológico de la proteína mutada. Una vez que se ha descrito un gen y su producto, el análisis de su secuencia puede emplearse para identificar secuencias homologas y motivos estructurales y funcionales con proteínas conocidas para un mejor entendimiento de sus actividades. Las interacciones funcionales de las proteínas son potencialmente significativas para estudiar el fenotipo cardíaco. Así, se ha establecido que la titina mutada (derivada de pacientes con HCM) reduce su afinidad de unión con otras proteínas sarcoméricas, como la α–actina, y se pueden caracterizar sus interacciones sinérgicas con factores de transcripción como el NKX2.5 y TBX5 en el desarrollo cardíaco temprano.65
Análisis transgénicos:
Los ratones transgénicos son una herramienta muy utilizada en biología molecular para confirmar un fenotipo específico, que puede ser modificado por la alteración de un gen y su producto proteico, en este tipo de organismos llamados "Knock–out" cuando se suprime el gen, o "knock–in" cuando se altera su expresión de manera positiva (sobre–expresión). Ratones knock–out para la oxidación de ácidos grasos (proteína trifuncional mitocondrial), la transcripción del mtDNA y la bioenergética (factor A de transcripción mitocondrial) y para el gen de la frataxina mitocondrial, desarrollan una rápida progresión de disfunción cardíaca y DCM con datos clínicos de cardiomiopatía asociada con mutaciones específicas en genes involucrados en la función bioenergética mitocondrial.66–68 Estas metodologías han proveído una gran cantidad de información para establecer el papel crítico que juega en gen TBX1 en la etiología del síndrome DiGeorge/Velocardiofacial. Ratones heterocigotos para un alelo TBX1 anulado presentan una gran incidencia de anormalidades en el tracto de salida cardíaco así como otras anormalidades características del síndrome DiGeorge.69
Análisis molecular de anormalidades fetales:
La reconstrucción tridimensional de los defectos cardíacos con la ayuda del ultrasonido, los rayos X y la resonancia magnética nuclear, ha impactado positivamente en el diagnóstico y las estrategias terapéuticas de las enfermedades cardíacas. Actualmente, la mayoría de enfermedades congénitas cardíacas pueden detectarse in útero con estas metodologías, de tal manera que se recomienda hacer evaluaciones subsiguientes de anormalidades extracardíacas y cromosómicas en los pacientes pediátricos, debido a que este tipo de anormalidades se presentan en un 62% y 38% de los casos, respectivamente. El consejo genético, basado en una evaluación prenatal, provee información real acerca de la incidencia, diagnóstico y pronóstico de los defectos cardíacos fetales. El diagnóstico prenatal de malformaciones cardíacas congénitas y sus correlaciones moleculares (por ejemplo: la microdeleción del cromosoma 22q11 en el Síndrome DiGeorge y del 7q en el síndrome de Williams), detectables por técnicas citogenéticas y moleculares subsecuentes a la amniocentesis, ha demostrado ser una potente herramienta en el manejo de las malformaciones del neonato como lo son la transposición de grandes arterias y el síndrome de hipoplasia cardíaca izquierda.
Enfermedades cardíacas adquiridas en niños:
Entre las enfermedades cardíacas adquiridas por neonatos y niños se encuentra la enfermedad de Kawasaki, enfermedad cardíaca reumática aguda y crónica, endocarditis infecciosa y miocarditis. La tecnología genética–molecular se ha utilizado en el diagnóstico temprano de estas enfermedades, aunque de forma limitada; sin embargo, su aplicación puede dar nueva información en el conocimiento general de las mismas. La enfermedad de Kawasaki es una vasculi–tis aguda limitada de la infancia o de la adolescencia temprana y es una de las principales causas de enfermedad cardíaca adquirida en Estados Unidos y Japón.70 Su etiología es desconocida, pues aún con las herramientas moleculares no se ha detectado alguna asociación viral o bacterial con la enfermedad. Si no es tratada, el 25% de los pacientes desarrollan aneurismas en las arterias coronarias. El tratamiento es efectivo si se aplica dentro de los primeros diez días de inicio de la enfermedad (para prevenir el involucramiento coronario), aunque esto es muy complicado para el cardiopediatra, pues debe distinguir la enfermedad de Kawasaki de otras enfermedades en un tiempo relativamente corto. En este sentido, el ultrasonido intravascular es una alternativa para este fin junto con marcadores moleculares que pueden usarse en forma de análisis por microarreglos para confirmar el diagnóstico.
Los análisis moleculares e inmunológicos tienen implícita la presencia de inducción viral (comúnmente se involucra el grupo B de los coxsackie virus o CVB) y respuestas autoinmunes aberrantes en la patogénesis de la miocarditis pediátrica, la cual, en algunos casos, puede evolucionar a DCM. En estudios moleculares recientes, con la ayuda de la PCR, se ha encontrado presencia de adenovirus, además de enterovirus, en el miocardio de niños con miocarditis y DCM.71 Por otro lado, aunque aún no se ha elucidado completamente el mecanismo patológico de la fiebre reumática o de la enfermedad cardíaca reumática inducida por Streptococus, los análisis moleculares han aportado avances importantes en los aspectos críticos autoinmunes de la enfermedad y se espera que futuros análisis de ligamiento y asociación genética generen información importante acerca de los factores genéticos involucrados en la susceptibilidad del paciente. Los datos moleculares también pueden utilizarse en diversas estrategias para el manejo de anormalidades cardiovasculares asociadas con infecciones adquiridas como la hipertensión pulmonar que se puede presentar por infección con HIV.72
Farmacogenética y cardioprotección:
El entendimiento de las enfermedades cardiovasculares a nivel genómico puede permitirnos hacer una mejor estratificación de subclases de pacientes para optimizar y dirigir terapias paciente–específicas.
Los campos relacionados con la farmacogenómica y con la farmacogenética abrazan la promesa de improvisar el desarrollo de drogas y la creación de terapias basadas en la capacidad individual de un organismo para metabolizar dichos compuestos tomando en cuenta la edad, influencia de la enfermedad, factores medioambientales (por ejemplo dieta), medicamentos tomados y factores genéticos individuales como la especificidad de transporte, metabolismo y blanco de la droga. Por ejemplo, un subgrupo de polimorfismos de base simple identificados en los genes humanos como el del receptor beta adrenérgico y la enzima convertidota de angiotensina (ACE), se han asociado con cambios substanciales en el metabolismo o efectos de medicamentos usados en el tratamiento de la enfermedad cardiovascular, y quizá sean informativos para predecir la respuesta clínica (Tabla III).73 La terapia individualizada puede ser crítica para establecer las dosis de las drogas y su eficacia en niños con enfermedades cardiovasculares, una población en la cual la fármaco–cinética de los medicamentos ha sido pobremente definida y muchas veces es impredecible. Los fenotipos inmunológicos y genéticos de los pacientes pediátricos pueden ser de gran ayuda en el establecimiento de estrategias terapéuticas más efectivas, tanto inhibiendo como estimulando respuestas específicas.
Un creciente número de evidencias han establecido que la cardioprotección puede ser evocada tanto por el preacondicionamiento isquémico o por mecanismos farmacológicos (por ejemplo nicorandil y diaxoxide) que pueden ser potencial–mente manejados como una estrategia de protección del órgano y sus tejidos en la enfermedad cardíaca isquémica o el insulto hipóxico, aunque a la fecha exista muy poca información concerniente a las respuestas cardioprotectoras en niños e infantes. Se han realizado modelos experimentales en animales de laboratorio que han establecido las bases moleculares de los mecanismos de cardioprotección, en los cuales se involucra una red de señales de traducción con diversas vías de señalización mediadas por receptores de superficie, activación y translocación subcelular de proteínas cinasas (como la PKC épsilon, P38 MAP cinasa y JUN cinasa) y la apertura de canales sarcolémicos y mitocondriales KATP.74 Se han encontrado niveles elevados de PKC épsilon, P38 MAP cinasa y JUN cinasa en infantes con defectos cardíacos cianóticos e hipoxia, las cuales no están activadas en infantes con defectos acianóticos o sujetos normales; esto indica que la ruta de señales de traducción cardioprotectoras se encuentran parcialmente en operación en infantes con hipoxia.75 En períodos breves de hipotermia, previos a un insulto isquémico prolongado, se ha observado cardioprotección asociada a las proteínas de estrés y a las señales mitocondriales y quizás éstas estén involucradas en el manejo clínico de la taquicardia ectópica por hipotermia.76,77 Futuras investigaciones en esta área podrán revelar moléculas blanco potenciales para una intervención farmacológica altamente específica. Sin embargo, como una nota precautoria, dichas investigaciones pueden llevarse mucho tiempo para incrementar nuestro conocimiento de la red de rutas de señalización que están interactuando. A pesar de los recientes logros en la identificación precisa de los defectos genéticos de señalización que causan arritmias cardíacas, el desarrollo de drogas efectivas (por ejemplo bloqueado–res de canales iónicos específicos), que puedan reducir sustancialmente la mortalidad asociada con severos desórdenes arrítmicos, se ha visto mermado y se incrementará en la medida en que nuestro conocimiento acerca de los complejos circuitos cardíacos, causas múltiples, factores genotípicos y de riesgo involucrados en estas enfermedades, aumente.78
Perspectivas:
Aunque se han realizado avances significativos en el diagnóstico y tratamiento de las enfermedades cardíacas en los niños, existen aún muchas preguntas por resolver a nivel de los mecanismos patofisiológicos básicos de dichos padecimientos. La aplicación de la tecnología genético–molecular ha trazado brechas por las cuales se han abordado las enfermedades cardiovasculares y han permitido el mapeo cromosomal y la identificación de algunos genes involucrados tanto en la etiología primaria como en los factores de riesgo para el desarrollo de dichas anomalías. Las siguientes áreas del conocimiento pueden ser muy promisorias en el futuro:
1) El entendimiento del desarrollo cardíaco y vascular ha sido poco explorado en los infantes, por lo cual la identificación futura de nuevos genes involucrados en la organogénesis y el desarrollo vascular servirá como un importante fundamento para el entendimiento de cómo se generan los defectos genéticos congé–nitos y sus fenotipos cardíacos. Los métodos bioinformáticos pueden emplearse para investigar las bases de datos generadas por medio de técnicas genéticas de rutina. Esto permitirá la subsiguiente clonación de nuevos genes a partir del cDNA de interés, con lo cual se puede lograr la caracterización temporal y espacial de patrones específicos de expresión genética de genes específicos del desarrollo embrionario (usando hibridación in situ).
2) Los mecanismos que gobiernan la especificación temprana de las cámaras cardíacas en el desarrollo del tubo cardíaco no se conocen totalmente, aunque se piensa que pueden estar involucradas nuevas rutas de señalización célula–célula entre las células emigrantes, así como el disparo de programas de expresión genética cámara–específicos, mediados por factores de transcripción especializados como la proteína morfogenética de hueso (MBP). Se pueden crear nuevas áreas para el estudio del papel de las moléculas de señalización (por ejemplo WNT) utilizando animales Knock–out (con gran variedad de trasfondos genéticos) y acceder a su interacción con factores de transcripción críticos como dHAND, NKX2.5, GATA4 y TBX. Algunas aproximaciones metodológicas semejantes pueden ser usadas para elucidar el sistema de conducción cardíaco y para descifrar el papel de los sistemas de señalización que participan en la formación vascular de células endoteliales, enfocándose en la interacción del VEGF, angiopoyetina, TGF y la ruta Notch.
3) Otra área crítica en la investigación cardiológica es la identificación de los reguladores moleculares que controlan la proliferación de los cardiomiocitos. Dichas células son mitóticamente activas durante la embriogénesis y generalmente cesa su proliferación antes del nacimiento. El estudio de las bases moleculares de la proliferación de los cardiomiocitos podría impactar positivamente en los intentos clínicos por reparar el tejido cardíaco dañado. Los mecanismos de regulación del crecimiento celular pueden ser investigados haciendo cuidadosas comparaciones entre los perfiles de expresión génica de miocitos embrionarios y de miocitos postnatales, así como por generación de cultivos de líneas celulares capaces de responder a estímulos inductores de proliferación. Alternativamente, el trasplante celular es un mecanismo utilizado para aumentar el número de miocitos en corazones dañados o isquémicos. Interesantemente, algunos estudios recientes han mostrado que una subpoblación de células troncales cardíacas de adulto inyectadas en un corazón isquémico fueron capaces de reconstituir completamente al miocardio diferenciado, formando tanto miocitos como vasos sanguíneos.79 De tal suerte, se deben realizar grandes esfuerzos en la investigación para definir las condiciones óptimas necesarias para la diferenciación y proliferación de los cardiomiocitos y para su completa integración funcional como células troncales en el miocardio, así como investigar la habilidad de las células troncales para reparar el tejido cardíaco en los niños con cardiomiopatía (por ejemplo DCM), enfermedad de Kawasaki con daño miocardico y ARVD, los cuales pueden ser ayudados por trasplante de células troncales. La visión dentro de las consecuencias cardiovasculares por una función genética anormal y su expresión, puede impactar sobre el desarrollo de estrategias terapéuticas dirigidas y manejo de enfermedades para los niños que presentan desórdenes cardíacos congénitos y adquiridos y quizá reemplacen a tratamientos menos efectivos dirigidos únicamente a rectificar los defectos cardíacos estructurales con mejoras funcionales temporales.
Desarrollo de cardiopatías congénitas cianóticas
Cianosis diferencial: la cianosis diferencial casi siempre indica alguna cardiopatía congénita; muchas veces persistencia del conducto arterioso y coartación de la aorta forman parte del complejo anatómico anormal. Cuando la parte superior del cuerpo es rosada y la inferior azul, es probable que exista coartación de la aorta o interrupción del arco aórtico, y existe sangra oxigenada en la parte superior del cuerpo y sangre insaturada, en la mitad inferior, que fluye del conducto arterioso de derecha a izquierda. Este último también sucede en pacientes con persistencia del conducto arterioso y elevación acentuada de la resistencia vascular pulmonar. En los pacientes con transposición de grandes arterias y coartación de la aorta con flujo retrogrado a través de una persistencia del conducto arterioso, la situación es la inversa la porción inferior del cuerpo es rosada y la superior de color azul, para confirmar la presencia de cianosis diferencial es útil valorar en forma simultanea la saturación de oxigeno en las arterias temporal o braquial derecha y femoral.
Cardiopatías congénitas cianóticas
Los pacientes con cardiopatías congénitas cianóticas tienen desaturación de oxigeno arterial ocasionada por cortocircuito de la sangre venosa sistémica hacia la circulación arterial o por anatomía cardiaca. La cual envía una mezcla de sangre de retorno sistémico y pulmonar. El cortocircuito puede ocurrir a nivel de las aurículas (CIA), ventrículos en la comunicación interventricular (CIV) o grandes vasos (PCA o ventana aortopulmonar). Si hay cortocircuito de derecha a izquierda implica que hay obstrucción del lado derecho, distal a ese nivel, o la presencia de enfermedad vascular obstructiva o pulmonar que causa inversión del flujo a través de una lesión previa de cortocircuito de izquierda a derecha.Así el cortocircuito de derecha a izquierda a través de una CIA (o agujero oval permeable) puede ser causado por atresia tricuspídea, estenosis tricuspídea, estenosis pulmonar grave o atresia con tabique ventricular intacto. O bien .enfermedad vascular pulmonar .también en la anomalía de ebstein (en ocasiones clasificada como cardiopatía acianótica) con insuficiencia tricuspídea grave y CIA asociada o agujero oval permeable .el cortocircuito de derecha a izquierda se debe al aumento de la presión en la aurícula derecha.
El cortocircuito de derecha a izquierda al nivel del ventrículo (a través de CIV) puede deberse a obstrucción del infundíbulo ventricular derecha creada por estenosis valvular o infundibular (tetralogía de fallot) o aumento de la resistencia vascular pulmonar (síndrome de eisenmenger). El cortocircuito de derecha a izquierda a través de un conducto permeable casi siempre se debe a enfermedad vascular pulmonar.
Desde el punto de vista fisiopatológico las cardiopatías con cortocircuito de derecha a izquierda se pueden dividir en tres grupos, cardiopatías obstructivas del lado derecho con comunicación al lado izquierdo del corazón, como ocurre con estenosis pulmonar severa asociada a CIV cardiopatías con mezcla total, en que retornos venosos sistémico y pulmonar se mezclan en una cavidad común como ocurre en un ventrículo único, y ; cardiopatías por falta de mezcla, con circuitos pulmonar y sistémico en paralelo, como ocurre en la transposición de grandes arterias.
| CARDIOPATIAS CONGENITAS CIANOTICAS (cortocircuito de derecha a izquierda) | Obstructivas Corazón Derecho | Tetralogía de Fallot, atresia pulmonar, ventrículo único o atresia tricuspídea con estenosis pulmonar |
| Mezcla Total | Ventrículo único o atresia tricuspídea sin estenosis pulmonar, truncus arterioso, drenaje venoso anómalo pulmonar total | |
| Falta de Mezcla | Transposición de Grandes Arterias |
En el segundo grupo se encuentran todas las cardiopatías complejas sin obstrucción al flujo pulmonar como atresia tricuspídea, ventrículo único, doble salida de ventrículo derecho, truncus arterioso, aurícula única, drenaje venoso anómalo pulmonar total no obstructivo. En este grupo al existir mezcla total entre retornos venosos pulmonar y sistémico sin obstrucción al flujo pulmonar, existe hiperflujo pulmonar marcado el que resulta en mayor retorno pulmonar que sistémico, por lo que la mezcla entre los retornos venosos resulta en saturaciones sistémicas sobre 80% e incluso en torno a 90%. Así las manifestaciones clínicas, además de la cianosis leve, son similares a las de las cardiopatías con cortocircuito de izquierda a derecha.
El tercer grupo corresponde a fisiología de transposición de grandes arterias, en que la falta de mezcla entre las circulaciones pulmonar y sistémica se produce porque la sangre desaturada que retorna por las venas cavas a la aurícula derecha vuelve a la aorta y circulación sistémica sin haber pasado por la circulación pulmonar, y la sangre oxigenada que retorna por las venas pulmonares a la aurícula izquierda, vuelve a dirigirse a los pulmones a través de la arteria pulmonar sin alcanzar la circulación sistémica. Así se producen dos circuitos independientes; uno sistémico con sangre desaturada que lleva a hipoxemia severa, y uno pulmonar con sangre oxigenada que no es utilizada. Obviamente esta situación es incompatible con la vida, salvo que exista algún nivel de mezcla entre las dos circulaciones, como el foramen oval y el ductus arterioso, donde se produzca cortocircuito bidireccional que permite mezcla entre las dos circulaciones y la sobrevida del neonato por al menos algunas horas. Desafortunadamente, la mezcla a estos niveles es frecuentemente insuficiente, y transitoria, ya que el ductus tiende a cerrarse en las primeras horas de vida y el foramen oval hace lo mismo en el curso de días a semanas. En estas cardiopatías el flujo pulmonar está normal o aumentado, pudiendo desarrollarse congestión pulmonar e incluso edema pulmonar.
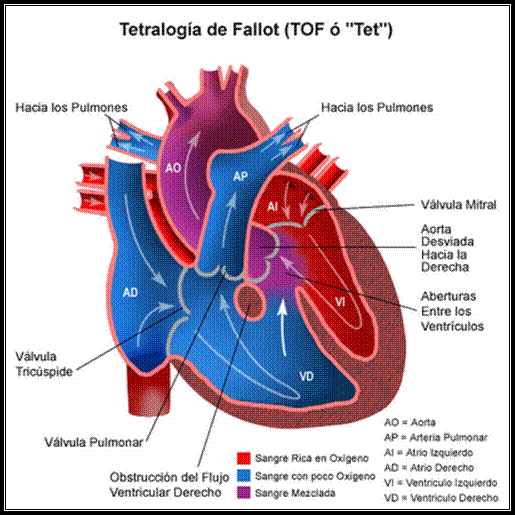
Tetralogía de Fallot
Bases para el diagnostico:- Antecedentes de intolerancia al ejercicio y posición en cuclillas durante la infancia
- Cianosis central, choque de punta prominente del ventrículo derecho. soplo de estenosis pulmonar (con flujo sanguíneo pulmonar prominente)y ausencia de ruido.
- Hipertrofia ventricular derecha leve; en ocasiones hipertrofia ventricular izquierda.
- La radiografía torácica muestra el corazón clásico en forma de bota (coer en sabot) en casos graves sin cortocircuito de izquierda a derecha; crecimiento de ventrículo izquierdo y dilatación posentenótica de la arteria pulmonar en casos más leves; arco aórtico en el lado derecho en casi 25 % de los pacientes.
- Los ecocardiogramas muestran hipertrofia ventricular derecha, cabalgamiento de la aorta. Grandes comunicaciones interventriculares perimenbranosas y obstrucción del infundíbulo ventricular derecho (subvalvulares, valvular supravalvular o en las ramas de la arteria pulmonar).
- Gradiente a través del infundíbulo ventricular derecho, presiones arteriales pulmonares normales, igualación de la presión en los ventrículos derecha e izquierda.
- Posibles ramas anómalas de arterias coronarias que cruzan el infundíbulo ventricular derecho, detectadas en la angiografía coronaria
La tetralogía de Fallot es la forma más común de cardiopatía congénita cianótica. Sin intervención quirúrgica, la mayoría de los pacientes fallece en la infancia; sin embargo, en ocasiones se encuentra en pacientes acianóticos con estenosis pulmonar moderada a leve y mínimo cortocircuito de derecha a izquierda (tetralogía de fallot rosada). Si bien se denomina tetralogía de fallot, sólo la CIV no restrictiva de tipo membranoso y la estenosis pulmonar contribuyen a la fisiopatología de este trastorno.
La gravedad de la estenosis pulmonar determina la presión sistólica del ventrículo derecho y por tanto el grado de cortocircuito de derecha izquierda. La estenosis pulmonar puede ser valvular o, con mayor frecuencia .infundibular con una banda muscular que causa obstrucción en el infundíbulo ventricular derecho los otros dos componentes de la tetralogía incluyen cabalgamiento aórtico e hipertrofia ventricular derecha secundaria. Tanto los pacientes cianóticos como acianótico se encuentran en lato riesgo de endocarditis, al igual que muchos pacientes con CIV complicada.
- Síntomas y signos
En casos graves de estenosis o atresia pulmonares, el paciente puede tener paliación quirúrgica que salve la vida con un cortocircuito aortopulmonar .en el pasado no se intentaron reparaciones totales hasta la edad escolar. Pero en muchos centros ahora se realizan en la lactancia.
El examen físico revela cianosis y, con menos frecuencia, dedos en palillos de tambor. El área precordial por lo general es silenciosa, si bien puede haber un soplo leve en ventrículo derecho. La intensidad y duración del soplo del flujo pulmonar, por lo general no hay ruido P2.en caso de atresia pulmonar con CIV (una forma extrema de tetralogía de fallot). Puede auscultarse un soplo continuo sobre la espalda por las colaterales aortopulmonares. Los pacientes que tienes una derivación "clásica" de Blalock-taussing, anastomosis de la arteria subclavia hacia un lado de la arteria pulmonar no tendrán pulso en el brazo ipsolateral y tendrán un soplo continuo en tanto la derivación este permeable y funcione en forma apropiada. Una derivación "modificada" de Blalock-taussing, interponiendo un tubo pequeño de Gore-tex entre la arteria subclavia y la arteria pulmonar, preserva el pulso humeral y se ocluye con facilidad al momento de la reparación intracardiaca. En pacientes que han tenido reparación intracardiaca, suele auscultarse un soplo pulmonar de tono bajo. Cuando el soplo ocurre sólo al inicio de la diástole, sugiere insuficiencia residual importante desde el punto de vista clínico.
- Estudios diagnósticos
Los datos radiográficos también dependen de la fisiopatología subyacente del individuo. El típico corazón en bota se observa cuando la estenosis pulmonar es grave y el ventrículo izquierdo es pequeño. En tales casos, se reduce el flujo sanguíneo pulmonar. La dilatación de la arteria pulmonar distal al sitio del a estenosis y del arco aórtico del lado derecho pueden ser visibles en la radiografía torácica.
2. Ecocardiografía: la ecocardiografia transtorácica bidimensional y doppler muestra las características de este defecto en su estado original y después de la reparación. Los datos típicos en pacientes con tetralogía de Fallot no reparada incluyen hipertrofia ventricular derecha grave y una válvula pulmonar malformada, engrosada con dilatación posentenótica. De otra forma, el nivel de la estenosis puede ser con predominio infundibular, con estrechamiento hipertrófico marcado del infundíbulo ventricular derecho.
3. Cateterismo cardiaco. El cateterismo cardiaco revela un gradiente a través del infundíbulo pulmonar, por lo general presión normal en arteria pulmonar e igualación de las presiones entre los ventrículos derecho e izquierdo. La angiografía puede definir mejor la anatomía del infundíbulo ventricular derecho y el tamaño de la comunicación interventricular .la arteriografía coronaria puede demostrar origen anómalo de la arteria coronaria izquierda.
4. Otros datos de laboratorio. La saturación arterial muestra reducción variable, y hay eritrocitocis secundaria en los adultos que no se han sometido a cirugía de reparación quirúrgica adecuada, la saturación arterial debe ser normal.
Pronóstico y tratamiento
Sólo 11% de los individuos que nacen con esta lesión sobreviven sin cirugía paliativa después de los 40 años. Como la estenosis pulmonar, casi siempre son elegibles para procedimiento quirúrgico en la edad adulta. Desde el punto de vista médico, es importante evitar el tratamiento sistémico con vasodilatadores en pacientes sin corrección de la tetralogía de fallot, porque una reducción en la presión arterial puede incrementar el cortocircuito de derecha a izquierda. La endocarditis es poco común en tetralogía de fallot no reparada.

Transposición de los grandes vasos
Es la cardiopatía mas frecuente de aquellas con aumento de la vascularización y cianosis .en la transposición completa encontramos una concordancia auriculoventricular, una discordancia auriculoventricular, de tal que la del ventrículo derecho sale la aorta y del izquierdo, la arteria pulmonar.La aorta nace en el ventrículo derecho y la arteria pulmonar en el ventrículo izquierdo. Este defecto es provocado por una anormal división del tronco arterioso por parte del tabique aorticopulmonar, el cual ha seguido una trayectoria recta en lugar de haberlo hecho en forma espiralada.
Se establecen así dos circulaciones en paralelo, entre la aurícula y el ventrículo derecho, pasando por todo el organismo a través de las arterias y venas sistémicas, y entre la aurícula y el ventrículo izquierdo a través de la circulación pulmonar. Si no se crea una circulación entre ambas, la lesión es incompatible con la vida. Generalmente existe un foramen oval permeable, pero suele ser insuficiente, de aquí la urgencia que existe en su diagnostico, para realizar a continuación una atrioseptomia paliativa (Rash-kind) que le permita mantenerse con vida hasta que le haga la intervención correctiva. Esta comunicación permite un flujo bidireccional de derecha a izquierda y viceversa, con lo que la aorta recibe parte de sangre oxigenada. A veces existe una amplia comunicación interauricular, interventricular o entre los grandes vasos, que evita la intervención paliativa.
En los primeros días de vida, el corazón y la circulación pulmonar pueden ser de tamaño normal. Para el radiólogo es importante saber que la transposición de los grandes vasos es el primer diagnostico a tener en cuenta en un recién nacido con cianosis, incluso con una silueta cardiovascular normal. Después del nacimiento, cuando aumenta el flujo pulmonar, aparece cardiomegalia con dilatación de las cámaras. Existe congestión pulmonar marcada, tanto activa como pasiva. En un 50% de los casos, el corazón presenta una forma oval o de huevo. Generalmente el segmento de la arteria pulmonar es cóncavo o plano, con estrechez del pediculo vascular superior (60%), ayudando por la ausencia del timo normal, que desaparece en estos pacientes (atrofia de stress).
La vascularización aparece aumentada, dependiendo del tamaño de la comunicación entre las circulaciones sistémicas y pulmonares. En el mediastino llama la atención de que el cayado aórtico no se visualiza en el contorno cardiaco izquierdo, debido a la posición mas medial de la aorta, y que el hilio izquierdo es más bajo que el derecho, al estar invertida la dirección del tronco de la arteria pulmonar, que además permite un flujo de la arteria pulmonar, que además permite un flujo preferencial hacia el pulmón derecho. La desviación de la aorta ascendente hacia la derecha produce una prominencia del borde superior derecho del mediastino. En proyección arterial, no es infrecuente que la parte alta de la silueta cardiaca esta ensanchada debido a la posición anterior de la aorta.
El estudio ecocardiografico bidimensional permite hacer el diagnostico, al demostrar la posición anterior de la aorta saliendo del ventrículo derecho y la arteria pulmonar del ventrículo izquierdo
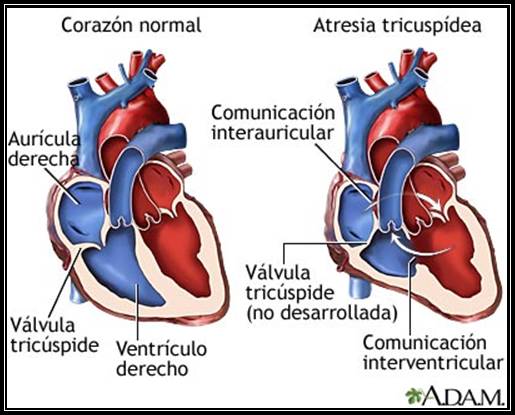
Atresia tricúspide
Atresia TricuspídeaEs un tipo de cardiopatía congénita en el cual la válvula tricúspide del corazón está ausente o no se ha desarrollado normalmente. Este defecto obstruye el flujo de sangre desde la aurícula derecha al ventrículo derecho del corazón.
Bases para el diagnóstico
- Antecedentes de cianosis (70%) o insuficiencia cardiaca congestiva (30%)
- Pacientes cianóticos con ausencia de choque de punta del ventrículo derecho y choque de punta prominente del ventrículo izquierdo.
- En la radiografía de tórax, campos pulmonares oligohemicos, prominencia de aurícula y ventrículos izquierdos sin crecimiento del ventrículo derecho en el espacio aéreo retroesternal.
- Datos de hipertrofia ventricular izquierda. válvula tricúspide atresica o ausente, comunicación interauricular, ventrículo derecho pequeño.
La atresia de la válvula tricúspide no puede observarse como una anomalía pequeña aislada .En tales pacientes, la válvula válvula tricúspide esta ausente, el ventrículo derecho se encuentra hiploplasico y la porción de entrada del ventrículo derecho se encuentra ausente. Si bien la comunicación auricular siempre esta presente, las anomalías asociadas adicionales determinan la fisiopatología fina y las manifestaciones clínicas.
La atresia Tricuspídea suele clasificarse de acuerdo con la presencia o ausencia de estenosis pulmonar y de transposición de aorta y arteria pulmonar.
Los grandes vasos tienen relaciones normales en casi 70% de los casos y muestran transposición 30%.
Casi todos los adultos con atresia Tricuspídea tienes cirugía previa y su estado clínico al momento de la presentación depende de la anatomía subyacente, así como lo adecuado de lo procedimiento paliativo.
Datos
A. Síntomas
§ Cianosis (coloración azulada de la piel)
§ Tendencia a la fatiga
§ Dificultad para respirar (disnea)
§ Dedos de las manos o de los pies en palillo de tambor
§ Insuficiencia cardiaca
§ Respiración rápida
§ Crecimiento deficiente
Signos y exámenes:
Esta afección se puede descubrir durante una ecografía prenatal de rutina o cuando el bebé es examinado justo después del nacimiento. La cianosis puede estar presente desde el nacimiento y generalmente se presenta un soplo cardíaco que puede aumentar su intensidad en algunos meses.
Los exámenes pueden ser los siguientes:
§ ECG
§ Ecocardiografía
§ Radiografía de tórax
§ Cateterismo cardíaco
§ IRM del corazón
Tronco arterial
CardiologíaEs un tipo raro de cardiopatía congénita, caracterizada por un solo vaso sanguíneo que sale desde los ventrículos derecho e izquierdo, en lugar de los dos vasos normales (la arteria pulmonar y la aorta).
Existen cuatro subtipos de tronco arterial, dependiendo de la anatomía específica del vaso único.
Tronco
Causas, incidencia y factores de riesgo
En la circulación normal, la arteria pulmonar procede del ventrículo derecho y la aorta procede del ventrículo izquierdo, los cuales están separados el uno del otro. Las arterias coronarias, que suministran sangre al músculo cardíaco, se desprenden de la aorta, justo por encima de la válvula ubicada a la entrada de esta arteria.
En el tronco arterial, un solo tronco arterial proviene de los ventrículos. Generalmente, también se presenta una comunicación interventricular grande (orificio entre los dos ventrículos). Como resultado, la sangre de color azuloso (no oxigenado) y la de color rojo (oxigenada) se mezcla por completo.
Parte de esta sangre mezclada va a los pulmones, otra parte a las arterias coronarias y el resto va al cuerpo. Generalmente, demasiada sangre es enviada a los pulmones. Entre tanto, la sangre que va a las arterias coronarias y al resto del cuerpo a menudo no contiene suficiente oxígeno.
Sin tratamiento, se pueden presentar dos problemas. Primero, los pulmones están llenos con líquido, lo que dificulta la respiración. El segundo problema es que los vasos sanguíneos que van a los pulmones se estrechan y resultan dañados de manera permanente. Con el tiempo, se le hace difícil al corazón bombearles sangre. Esto se denomina hipertensión pulmonar y puede ser mortal.
El tronco arterial es muy poco común.
Síntomas
- Insuficiencia cardíaca
- Letargo
- Mala alimentación
- Dificultad para respirar (disnea)
- Respiración rápida (taquipnea)
- Fatiga
- Cianosis (coloración azulada de la piel)
- Crecimiento lento o retraso en el crecimiento
- Ensanchamiento de las puntas de los dedos de las manos (dedos hipocráticos)
El cardiólogo o el pediatra generalmente perciben un soplo cuando escuchan el corazón con un estetoscopio.
- Un ECG muestra signos de agrandamiento del corazón (hipertrofia ventricular).
- Una radiografía del tórax evidencia agrandamiento del corazón y pulmones llenos de líquido.
- Una ecocardiografía muestra una comunicación interventricular y el diagnóstico definitivo de una sola arteria troncal.
- Rara vez, se necesita un cateterismo cardíaco para ayudar en el diagnóstico o planear una estrategia de tratamiento.
- IRM del corazón.
Se necesita cirugía para tratar esta afección. Hay dos procedimientos disponibles. Uno es el cerclaje de las arterias pulmonares que nacen en el tronco, pero ya se utiliza muy poco. El otro procedimiento se denomina reparación completa que parece ser la opción preferida pero, a medida que crece el niño, puede ser necesario repetir los procedimientos quirúrgicos.
Expectativas (pronóstico)
La reparación completa generalmente produce buenos resultados y es posible que sea necesario repetir la operación a medida que el paciente crece. Los casos que no reciben tratamiento tienen un pronóstico desalentador y generalmente llevan a la muerte durante el primer año de vida. Rara vez, el diagnóstico se pasa por alto hasta el comienzo de la vida adulta; por lo general, estos pacientes necesitan un trasplante de corazón y de pulmón.
Complicaciones
- Insuficiencia cardiaca
- Hipertensión pulmonar (presión arterial alta en los pulmones) con enfermedad pulmonar obstructiva
La persona debe consultar con el médico si su bebé o su hijo parece letárgico, no se alimenta bien, parece excesivamente cansado o tiene una leve dificultad para respirar o si parece no estar creciendo o desarrollándose de manera normal.
Si la piel, los labios o los lechos ungueales del niño se tornan azules o si el niño parece tener dificultad para respirar, se debe llevarlo a la sala de emergencias o hacerlo examinar con prontitud.
Corazón derecho hipoplásico
DEFINICIONCardiopatía cianótica compleja en la que existe una sola cámara ventricular, habitualmente la izquierda, por estenosis o atresia tricúspide o pulmonar, siendo mas frecuente la atresia tricúspide. Sin embargo, una estenosis pulmonar grave puede progresar in útero a atresia pulmonar con hipoplasia de ventrículo derecho.
INCIDENCIA
Las atresias tricúspide y pulmonar constituyen alrededor de 2% de las cardiopatías congénitas que se observan después del nacimiento. En series fetales, la atresia tricúspide comprende el 4%, y la estenosis y atresias pulmonares el 5% de todas las lesiones.
EMBRIOLOGIA
Pareciera que cualquier disminución del flujo sanguíneo puede conducir a desarrollo insuficiente de una cavidad en útero. Sin embargo, la atresia tricúspide podría ser causada por obliteración temprana del orificio ventricular derecho.
DIAGNOSTICO PRENATAL
Se puede realizar diagnostico antenatal mediante la realización de ecografía fetal. En el corazón fetal normal, los ventrículos tienen tamaño similar, por lo tanto la desproporción de tamaño entre los ventrículos orienta al diagnostico de hipoplasia por agenesia o atresia tricúspide. Además, se pueden visualizar malformaciones asociadas como: obstrucción arco aórtico, transposición de grandes arterias o estenosis pulmonar.
FORMAS DE PRESENTACION CLINICA
Habitualmente hay síntomas dentro de posprimeros días o semanas de vida . El recién nacido presenta cianosis, cuya intensidad va a depender de la magnitud del flujo pulmonar. Si el foramen oval es restrictivo habrá además disminución del débito cardíaco, con hipoperfusión sistémica evidencias de falla cardiaca derecha y congestión venosa sistémica (hepatomegalia e ingurgitación yugular).
A menor flujo pulmonar, mayor cianosis, que podrá presentarse desde el período de recién nacido, pero la mayoría de las veces su expresión clínica semejante a la del Fallot, incluso con crisis anoxénicas provocadas por estenosis infundibular. En la atresia tricúspide con flujo pulmonar disminuido, es infrecuente la Insuficiencia cardiaca, pero puede aparecer si la CIA es restrictiva.
Si además de la atresia tricúspide hay atresia pulmonar, la obstrucción será doble con hipertensión de aurícula derecha y schunt de derecha a izquierda auricular, el flujo pulmonar será ductus dependiente, el recién nacido tendrá cianosis acentuada y soplo sistólico moderado de insuficiencia tricúspide y soplo continuo ductal.
MALFORMACIONES ASOCIADAS
Este frecuente encontrar transposición de los grande vasos, atresia o estenosis pulmonar, dextrocardia, isomerismo auricular derecho, canal A-V, drenaje pulmonar anómalo total. Aunque las anormalidades cromosómicas son relativamente raras en presencia de lesiones valvulares del lado derecho, existirán en 5% de los fetos con atresia pulmonar y en 2% con atresia tricúspide.
Se debe realizar cariotipo en casos de ventrículo derecho hipoplásico, pues se han informado malformaciones extracardiacas en 34% de los fetos con atresia tricúspide.
Anomalía de Ebstein de la válvula tricúspide
La Anomalía De Ebstein de la válvula tricúspide es un defecto raro en el cual están desplazadas las inserciones de la válvula tricúspide hacia el ventrículo derecho en grandes variables. La anomalía de ebstein comprende una gama de anomalías que incluyen cierto grado de desplazamiento de la válvula tricúspide, de tamaño variable del ventrículo derecho y obstrucción variable de salida del flujo pulmonar. Las anomalías concurrente son una atrial septal defects (ASD), atresia pulmonar y transposición corregida congénitamente. Las hojuelas posterior y septal de válvula tricúspide están desplazadas en grado variable hacia el vértice del ventrículo derecho. Ello da por resultado una porción auriculizada del ventrículo derecho.La hojuela anterior permanece grande y parecida a una vela. El principal problema hemodinámico es incompetencia Tricuspídea con disminución del flujo sanguíneo pulmonar, y cuando existe un ASD, cortocircuito de derecha a izquierda que causa cianosis.
La incompetencia Tricuspídea de larga duración da lugar a sobrecarga de volumen de un ventrículo derecho anormal. La obstrucción variable del infundíbulo del ventrículo derecho limitara el flujo sanguíneo pulmonar adecuado, se asegura la presentación neonatal.
Diagnostico e intervención.Las formas mas graves de anomalía de ebstein se manifiestan con cianosis en la infancia. Los recién nacidos tienden a presentar una forma grave de la afección, con un ventrículo derecho notablemente ineficiente complicado por la resistencia pulmonar. La mortalidad en este grupo es alta. Los pacientes mayores presentan insuficiencia cardiaca y pueden tener cianosis. Las arritmias supraventriculares y el síndrome de preexcitacion (Wolf parkinson White ) se acompañan de anomalía de ebstein . la ecocardiografia es diagnostica. Los recién nacidos graves tienen índices de supervivencia malos y esta indicada la cirugía solo después de estabilizar con PGE1 y ventilación controlada. En el paciente de mayor edad, la cianosis e insuficiencia cardiaca son indicaciones para intervenir.
Ventrículo único:
Esta designación alude a un conjunto de lesiones complejas en el que ambas válvulas auriculoventriculares o una válvula aurículo ventricular común se abren a una única cavidad ventricular.Las anomalías asociadas son frecuentes y comprenden posiciones anormales de las grandes arterias, estenosis valvular o subvalvular de la arteria pulmonar y estenosis subaortica .
La supervivencia hasta la edad adulta depende de la existencia de un flujo palomar relativamente normal aunque con resistencia pulmonar también normal y de una buena función ventricular. Para estos enfermos se usa casi siempre la técnica de Fontan modificada, que consiste en crear una o varias vías de paso desde las vías sistémicas a las arterias pulmonares
Cardiopatía congénita
 |
 |
Definición
La enfermedad cardíaca congénita o cardiopatía congénita se refiere a problemas con la estructura y funcionamiento del corazón debido a un desarrollo anormal de éste antes del nacimiento. Congénito significa presente al nacer.
Nombres alternativos
Causas, incidencia y factores de riesgo
La cardiopatía congénita (CPC) puede describir muchos problemas diferentes que afectan al corazón y es el tipo de anomalía congénita más común. La cardiopatía congénita es responsable de más muertes en el primer año de vida que cualquier otro defecto de nacimiento. Muchos de estos defectos necesitan un seguimiento cuidadoso; algunos se curan con el tiempo, mientras que otros requerirán tratamiento.
La cardiopatía congénita suele estar dividida en dos tipos: cianótica (coloración azulada producto de una relativa falta de oxígeno) y no cianótica. Las siguientes listas cubren las cardiopatías congénitas más comunes:
Cianóticas:
Para la mayoría de defectos cardíacos congénitos no se puede identificar una causa conocida y se continúan haciendo investigaciones acerca de este tipo de cardiopatías. Fármacos como el ácido retinoico para el acné, sustancias químicas, el alcohol e infecciones (como la rubéola) durante el embarazo pueden contribuir a algunos problemas cardíacos congénitos.
La cardiopatía congénita suele estar dividida en dos tipos: cianótica (coloración azulada producto de una relativa falta de oxígeno) y no cianótica. Las siguientes listas cubren las cardiopatías congénitas más comunes:
Cianóticas:
- Tetralogía de Fallot
- Transposición de los grandes vasos
- Atresia tricúspide
- Drenaje venoso pulmonar anómalo total
- Tronco arterial
- Corazón izquierdo hipoplásico
- Atresia pulmonar
- Algunas formas de drenaje venoso pulmonar anómalo total
- Anomalía de Ebstein
- Comunicación interventricular (CIV)
- Comunicación interauricular (CIA)
- Conducto arterial persistente (CAP)
- Estenosis aórtica
- Estenosis pulmonar
- Coartación de la aorta
- Canal auriculoventricular (defecto de relieve endocárdico)
Para la mayoría de defectos cardíacos congénitos no se puede identificar una causa conocida y se continúan haciendo investigaciones acerca de este tipo de cardiopatías. Fármacos como el ácido retinoico para el acné, sustancias químicas, el alcohol e infecciones (como la rubéola) durante el embarazo pueden contribuir a algunos problemas cardíacos congénitos.
Síntomas
Los síntomas dependen de la afección específica. Aunque la cardiopatía congénita está presente al nacer, los síntomas pueden no ser obvios inmediatamente. Defectos como la coartación de la aorta pueden no causar problemas durante muchos años. Otros problemas, como una comunicación interventricular (CIV) pequeña, pueden no causar nunca ningún problema y algunas personas con esta afección pueden tener actividad física y un período de vida normales.
Signos y exámenes
Los exámenes de diagnóstico dependen de la afección específica.
Tratamiento
El tratamiento depende de la afección específica. Algunas cardiopatías congénitas se pueden tratar sólo con medicamentos, mientras que otras requieren una o más cirugías.
Grupos de apoyo
Expectativas (pronóstico)
El pronóstico del paciente depende del defecto específico.
Complicaciones
Las complicaciones dependen de la afección y tratamiento específicos.
Situaciones que requieren asistencia médica
Consulte con el médico si sospecha que su hijo tiene un problema cardíaco.
Prevención
Evite el consumo de alcohol y otras drogas durante el embarazo. A los médicos se les debe hacer saber que una mujer está embarazada antes de prescribirle algún medicamento. Se debe realizar un examen de sangre a comienzos del embarazo para ver si la mujer tiene inmunidad contra la rubéola. Si la madre no tiene inmunidad, tiene que evitar cualquier exposición posible a esta enfermedad y debe ser vacunada inmediatamente después del parto.
Los niveles de azúcar en la sangre mal controlados en mujeres con diabetes durante el embarazo también están asociados con una alta tasa de defectos cardíacos congénitos durante este período.
Los expertos creen que algunos medicamentos que necesitan receta y otros de venta libre, al igual que las drogas psicoactivas durante el embarazo, aumentan el riesgo de desarrollo de defectos cardíacos.
Asimismo, puede haber algunos factores hereditarios que jueguen un papel en el desarrollo de las cardiopatías congénitas. La genética en verdad parece jugar un papel en muchas enfermedades y muchos miembros de la familia pueden resultar afectados. Hable con el médico acerca de la realización de pruebas de detección.
Las madres gestantes deben recibir un buen cuidado prenatal. Muchos de estos defectos se pueden descubrir en ecografías de rutina llevadas a cabo por un obstetra. Entonces, se puede anticipar el parto y el personal médico adecuado (como un cardiólogo pediatra, un cirujano cardiotorácico y un neonatólogo) puede estar presente y listo para ayudar en la medida de lo necesario. Dicha preparación puede significar la diferencia entre la vida y la muerte para algunos bebés.
Los niveles de azúcar en la sangre mal controlados en mujeres con diabetes durante el embarazo también están asociados con una alta tasa de defectos cardíacos congénitos durante este período.
Los expertos creen que algunos medicamentos que necesitan receta y otros de venta libre, al igual que las drogas psicoactivas durante el embarazo, aumentan el riesgo de desarrollo de defectos cardíacos.
Asimismo, puede haber algunos factores hereditarios que jueguen un papel en el desarrollo de las cardiopatías congénitas. La genética en verdad parece jugar un papel en muchas enfermedades y muchos miembros de la familia pueden resultar afectados. Hable con el médico acerca de la realización de pruebas de detección.
Las madres gestantes deben recibir un buen cuidado prenatal. Muchos de estos defectos se pueden descubrir en ecografías de rutina llevadas a cabo por un obstetra. Entonces, se puede anticipar el parto y el personal médico adecuado (como un cardiólogo pediatra, un cirujano cardiotorácico y un neonatólogo) puede estar presente y listo para ayudar en la medida de lo necesario. Dicha preparación puede significar la diferencia entre la vida y la muerte para algunos bebés.
No hay comentarios:
Publicar un comentario